
為了 2050 淨零排放的目標,太陽能發電為不可或缺的再生能源之一,其中「鈣鈦礦太陽能電池」是近年最熱門的研究領域,不僅成本低廉、光電轉換效率也可達到 25%。然而,鈣鈦礦材料在環境中容易降解,影響使用壽命。材料科學家為了做出效能好又穩定的鈣鈦礦「料理」,無不卯足了勁,替這道菜加上各種「食材」,但是越複雜的菜,調出好味道就越困難。人腦畢竟有限,如果交給機器呢?中央研究院「研之有物」專訪院內應用科學研究中心包淳偉研究員,他與團隊訓練了一套機器學習模型,可以又快又準的找出複雜鈣鈦礦材料的最佳化條件!
光電好夥伴:複雜鈣鈦礦材料
對太陽能電池來說,鈣鈦礦材料具有優異的光電性質和低生產成本,近年也廣泛應用在 LED、雷射、光感測器和光觸媒。
鈣鈦礦是什麼呢?最初是指鈣與鈦的氧化物 CaTiO3,而現在常講的「鈣鈦礦材料」為一種統稱,泛指擁有相似結構的金屬鹵化物材料,通式為 ABX3。要調配出優秀的鈣鈦礦材料並不容易,科學家必須像大廚一樣,運用各種「食材」煮出 ABX3。
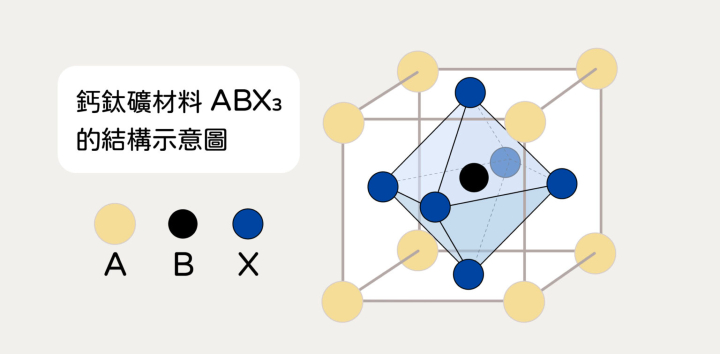
▲ 鈣鈦礦材料 ABX3 的結構示意圖,同一個位置可以放入不同的相應元素。(Source:Journal of Energy Chemistry,研之有物整理)
由於鈣鈦礦材料在環境中容易降解、影響使用壽命。研究發現,添加多種有機和無機離子的鈣鈦礦太陽能電池可大幅提升性能和穩定性,因此科學家為了調配出最好的鈣鈦礦材料,加料不手軟,成份也愈來愈複雜。
在眾多複雜鈣鈦礦材料中,包淳偉研究員探討的是 MAyFA1−yPb(BrxI1−x)3,下標符號 y 和 1-y 表示相對含量,如果 MA 佔 60%、FA 就是 40%,因為 MA 和 FA 會競爭同一個位置;同理 Br 和 I 亦然。

▲ 圖為鈣鈦礦材料通式 ABX3 對應到混合離子鈣鈦礦材料 MAyFA1−yPb(BrxI1−x)3 之示意圖。
問題來了,MAyFA1−yPb(BrxI1−x)3 這個材料這麼複雜,比例要怎麼配比較好呢?「你累積的經驗越多,就猜得越準」,包淳偉說。
2016 年曾經有國外團隊為了找出離子濃度配方與 MAyFA1−yPb(BrxI1−x)3 元件性能的關係,不惜花重本「土法煉鋼」,分別將兩組相對含量 7 等分(0, 1/6, 2/6, 3/6, 4/6, 5/6, 1),做出 49 種不同的鈣鈦礦太陽能電池,再去測量光電轉換效率,得出最佳比例為 MA2/6FA4/6Pb(Br1/6I5/6)3 。
然而,為何這樣的濃度配方可以得到最佳元件呢?很遺憾的,實驗團隊由於實驗表徵手段的限制,並不能解答這個重要的基礎問題。因此,實驗團隊仍然需要學生們焚膏繼晷的爆肝,用試誤法(trial and error)把最佳配方「踹」(try)出來。
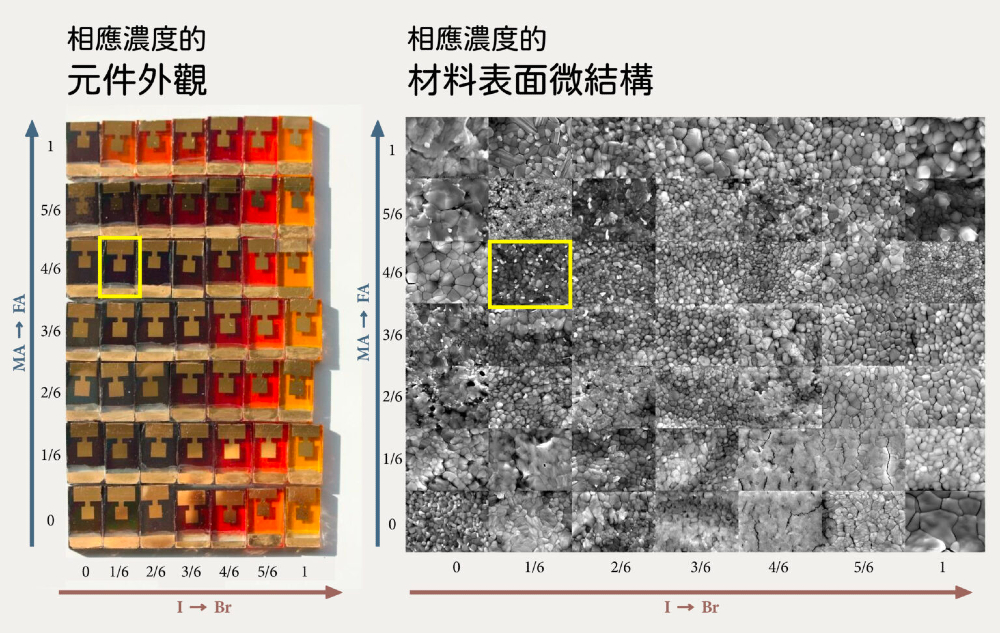
▲ 國外團隊為了找到 MAyFA1−yPb(BrxI1−x)3 最佳比例,做出 49 種不同的鈣鈦礦太陽能電池,黃框處即為最佳比例。左圖為相應濃度的元件外觀,右圖為相應濃度的材料表面微結構。(Source:Energy & Environmental Science)
不過,一直反覆試誤並非好方法,畢竟每做一次實驗就是一次成本。因此,科學家也設法從理論模擬著手,包淳偉強調「模擬的好處是可以在電腦空間中創造一個最純淨的系統」,而原子尺度模擬,更可以達到原子級的解析度,提供許多實驗無法量測的資訊。
材料科學注重製程(Process)、性質(Property)和結構(Structure)之間的關係。當我們對結構不夠了解時,往往只能透過不同的製程參數,慢慢做出我們想要的性質,可能在失敗多次之後,才能抓到一些訣竅。
理論模擬幫助科學家在做出樣品之前,先建立能量模型,找出能量最低、最穩定的微結構。當我們了解結構之後,可以避免有問題的製程參數設定,進而得到較好的材料性質。
首先,如果要知道材料性質,有個最精準也最耗時的方法:「第一原理計算」,只用量子力學原理,從頭開始把原子間的作用力和能量計算出來。
因為計算繁瑣,應用上只能模擬 1 奈米以內(10-9 公尺)的三維材料,抓到數個皮秒(10-12 秒)內的原子狀態,若再往外擴展所耗費的時間和成本難以想像。
相對地,計算材料性質也有省時省力的方法:「分子動力學模擬」,運用古典的牛頓力學,搭配統計力學去計算系統的微觀結構和能量。
分子動力學模擬大約可以模擬 100 奈米內的三維材料,抓到數個微秒(10-6 秒)內的原子狀態,可模擬的系統尺寸和時間都比第一原理計算要來得多!可惜準確度對於現在化學組成高度複雜的新穎材料而言是一個極大的挑戰。
有沒有一種方法,可以做到又快又準呢?有有有!它就是近年大熱門的「機器學習」!

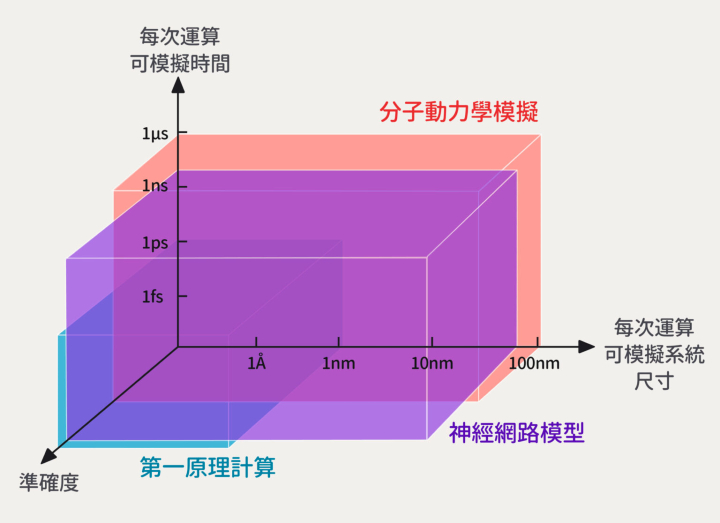
▲ 第一原理計算僅適合用在 1 奈米以內尺度,計算準確耗時;分子動力學模擬可用於 100 奈米尺度,計算省時卻不夠精準;透過機器學習建立的神經網路模型,可以快速模擬 100 奈米尺度的材料,也保留高準確度。(Source:包淳偉,研之有物整理,下同)
當包淳偉看到 2016 年國外團隊的 MAyFA1−yPb(BrxI1−x)3 鈣鈦礦研究之後,他認為「結構」這塊還有很多地方可以討論,如果透過理論模擬,先找出最低能量的微結構,或許就能更有效率地探索離子濃度空間,找出決定最佳配方的關鍵要素!
由於第一原理計算和分子動力學模擬都不夠好用,包淳偉就將念頭轉到近年熱門的「機器學習」,他和團隊就先從簡單的 PbI2 開始,慢慢做到複雜的鈣鈦礦材料。一開始包淳偉的團隊使用布朗大學開發的原子尺度機器學習套件(Atomistic Machine-learning Package,AMP)來進行訓練與測試,然而,由於 AMP 套件性能無法達到預期,包淳偉團隊就走上了自行開發機器學習分子動力學模擬程式的不歸路。
訓練神經網路模型時,包淳偉採用第一原理計算的結果當作機器學習素材,並設計函數進行反饋校正,直到預測的原子能量誤差遠小於熱擾動。
這套神經網路模型如何運作?先輸入原子座標(位置向量 r),再換算成「原子指紋」(特徵向量 G,表示該原子與其他原子之間獨一無二的相對關係),之後透過神經網路,快速輸出整個材料系統的原子能量和作用力。
從輸入到輸出,要模擬原子走一個步階(註 1)有多快?假設以 2,000 顆原子的計算量來看,自行開發的機器學習方法只要約 0.1 秒,第一原理計算則要花費 3 小時,足足快了十萬倍(註 2)!

▲ 包淳偉與團隊成功訓練出可以模擬複雜鈣鈦礦材料系統的神經網路模型。
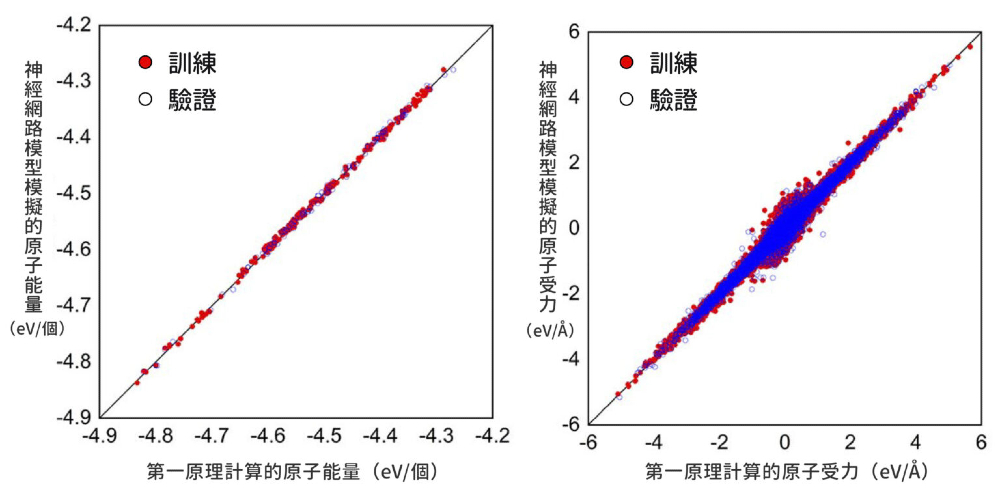
▲ 此神經網路模型可以準確預測 MAyFA1−yPb(BrxI1−x)3 鈣鈦礦材料的系統能量和受力。縱軸表示包淳偉團隊的神經網路模型模擬結果,橫軸表示第一原理計算結果。
包淳偉團隊成功訓練出來的神經網路模型,可以在 2,000 顆原子左右的材料系統上進行數百萬種可能的原子排列採樣,並計算出複雜鈣鈦礦材料的最低能量結構,模擬出不同原子在材料中最穩定的位置、它們的振動,以及它們受到擠壓時會怎麼跑。
多虧了神經網路的快速計算,即使是 MAyFA1−yPb(BrxI1−x)3 這麼複雜的系統也能處理,跑了將近 100 萬次結構模擬,得出不同成份比例下 81 種最低能量的微結構(如下圖),這是第一原理計算絕對跑不出來的成果。
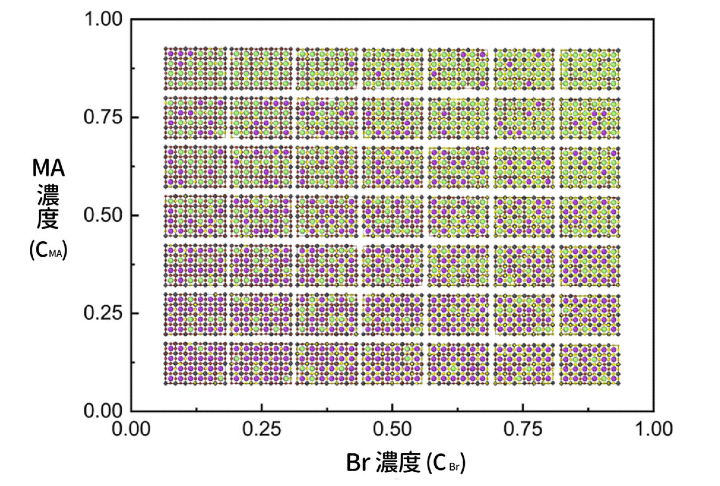
▲ MAyFA1−yPb(BrxI1−x)3 鈣鈦礦材料的最低能量原子結構,縱軸 y 為 MA 濃度(CMA,從 MA0-FA1到 MA1-FA0),橫軸 x 為 Br 濃度(CBr,從 Br0-I1 到 Br1-I0),各自 9 等分。為求圖片簡潔,省略 x, y = 0 或 1 的結構圖。
找出系統最低能量的原子組態還不夠,包淳偉團隊想要進一步檢驗鈣鈦礦材料 MAyFA1−yPb(BrxI1−x)3 是否能穩定地保持混合狀態,因此計算不同濃度成份下的離子混合能 Emix(如下圖)。
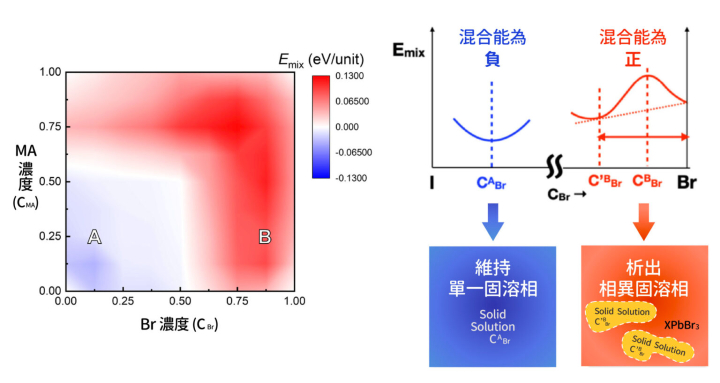
▲ MAyFA1−yPb(BrxI1−x)3 鈣鈦礦材料的混合能 Emix分布,藍色表示混合能為負(維持單一固溶相),紅色表示混合能為正(析出相異固溶相),可以看到 Br 和 MA 濃度高的時候,容易析出化合物。其中,縱軸 y 為 MA 濃度(CMA),橫軸 x 為 Br 濃度(CBr)。
從 MAyFA1−yPb(BrxI1−x)3 混合能分布初步來看,Br 濃度(CBr)或 MA 濃度(CMA)越高的時候,混合能就越高,系統越容易析出相異的固溶相。
除了混合能之外,研究團隊更進一步檢驗了不同濃度成份下的其他結構參數,例如短程有序參數 αA-B(正值表示 A-B 析出;負值表示 A-B 混合)、晶格扭曲 ηs(shear strain)與晶格畸變 ηv(volumetric strain),觀察析出化合物時,是否真的會改變晶格的幾何結構。
為了將模擬結果和實際情況對照,包淳偉再將模擬出來的結構以第一原理計算出不同濃度成份下的材料能隙(Eg),以及用內差法比對 2016 年國外團隊的實驗數據,得出不同濃度成份下的元件短路電流(Jsc)和光電轉換效率(power conversion efficiency, PCE)。
有了這些關鍵數據,我們終於可以完成鈣鈦礦材料 MAyFA1−yPb(BrxI1−x)3 最佳化製程參數的最後一哩路!
還記得我們一開始跑模擬的目標嗎?幫助研究團隊在花大錢做實驗之前,先找出最穩定的結構,從結構參數回推好的製程參數,進而得到較好的材料性質。
那麼要如何把這麼多參數的相關性一網打盡呢?有個好工具叫「皮爾森相關性矩陣」(Pearson correlation matrix)。


